

 Previous page
Previous page
Enhanced efficiency of numerically exact simulations in MAS NMR (M. Bechmann, S. Dusold, X. Helluy, and A. Sebald)
The vast majority of MAS Nuclear Magnetic Resonance (NMR) spectra cannot be analysed by empirical inspection. Instead, numerically exact simulations are necessary in order to determine unknown parameters from experimental MAS NMR spectra. Unfortunately, conventional numerically exact simulations of MAS NMR spectra are, in most cases, very time-consuming, to the extent that in-depth analysis is often not feasible for reasons of computing time. Furthermore, such simulations are not only necessary for the analysis of experimental spectra, but also for the evaluation and selection of the most suitable experimental pulse sequence for a given task. Owing to the enormous importance of efficient numerical simulation tools for the successful application of MAS NMR experiments, a major effort has been devoted to the improvement of the efficiency of our simulation routines. So far, emphasis has been placed on two particular aspects:
1) MAS NMR spectra of polycrystalline powders represent the superposition of spectra originating from very large numbers of small, randomly oriented 'single crystals'. Hence, one time-consuming step in the simulation procedures is the calculation of powder averages. This calculation step can be enormously accelerated by employing parallel-computing methods.
2) Further potential in saving CPU time lies in the computational methods used to calculate the time evolution of the spin dynamics. Magic-angle spinning itself imposes periodicity on the Hamiltonian, but most pulse sequences are not cyclic and periodic with the MAS rotation period. While brute-force integration ('direct method') in small time increments is generally possible, this calculation method is far too slow, even for the most simple spin systems and/or pulse sequences. Computational methods that take advantage of inherent periodicity and only require to carry out the calculation over the duration of a single rotation period (so-called COMPUTE methods) are much more efficient but are not generally applicable. A practical compromise which still offers major savings in CPU time is an approach using the 'direct method' only where absolutely necessary and switching to COMPUTE methods where possible ('mixed method').
Typical times required for the calculation of a single powder spectrum are quoted in the table below and hold for a Linux cluster consisting of eight dual-processor 450 MHz PCs. We believe to have one of the worldwide most efficient, generally applicable tools for purposes of MAS NMR simulations, running on economic PC hardware. The results reported further below all take advantage of these numerical tools.
Table 1: Typical times t [s] required for the calculation of R2-DQF (see sections ii)-iv)) MAS NMR spectra
| t(2-spin system) | t(3-spin system) | t(4-spin system) | |
| 'direct method', single processor | 8016 | not determined | not determined |
| 'mixed, method', single processor | 291 | 1454 | 9395 |
| 'mixed method', parallel processors | 23 | 100 | 654 |
Evaluation and comparison of double-quantum filtration techniques at the rotational-resonance condition in the presence of large shielding anisotropies (M. Bechmann, S. Dusold, X. Helluy and A. Sebald)
By means of exciting double-quantum coherences in MAS NMR it becomes possible to observe selectively coupled sub-sets of spins in the presence of (unwanted) background resonances as well as within large sets of coupled spins. Often the ultimately wanted information is encoded in the magnitudes and orientations of chemical shielding tensors.
The presence of large chemical shielding anisotropies, however, strongly degrades the efficiencies of many currently known pulse sequences that are suitable, in principle, for double-quantum filtration (DQF) experiments under MAS NMR conditions. For reasons of selectivity of the MAS DQF experiments, we have investigated the performance of several such NMR pulse sequences applicable at the so-called rotational-resonance (R2) condition, in the presence of large chemical shielding anisotropies.
We find, for instance, that a relatively simple two-pulse sequence performs best under these conditions in terms of R2-DQF efficiencies, and in addition yields MAS NMR lineshapes which can be fully and quantitatively analysed by numerical simulations. As an example, some experimental and simulated 13C R2-DQF MAS NMR spectra of the 13C spin pair in selectively 13C1,13C2-enriched solid sodium pyruvate are shown in Figure 3.7-6.
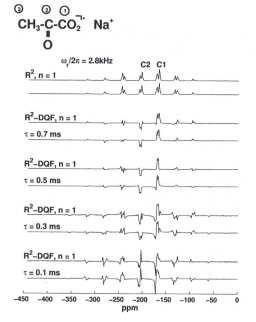 |
R2-DQF experiments on larger-than-two-13C spin systems in isotopically enriched materials (M. Bechmann, X. Helluy and A. Sebald)
For the general applicability and usefulness of R2-DQF MAS NMR experiments it is important that these experiments can also be performed on larger-than-two-spin systems. Consider, for example, the large coupled 31P spin systems in inorganic phosphates, where the 100 % naturally abundant isotope 31P cannot be depleted. Here we present first results on a 13C three-spin system in the biologically important phosphoenolpyruvate moiety. Note that the simulated and experimental 13C R2-DQF MAS NMR spectra of U13C-(NH4)3(PEP) (see Fig. 3.7-7) fully describe the molecular geometry of the phosphoenolpyruvate anion.
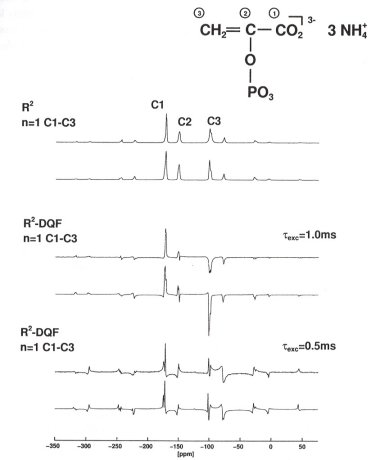 |
R2-DQF experiments on spin-1/2 isotopes in low natural abundance (X. Helluy and A. Sebald; in collaboration with C. Marichal/Mulhouse)
Another important feature of R2-DQF MAS NMR experiments is a sufficient efficiency of the double-quantum excitation and reconversion steps in the pulse sequence, in order to make these experiments feasible for spin-1/2 isotopes in low (natural) abundance. As a practical example, consider the network of Si sites in an inorganic silicate phase: 29Si isotopic enrichment would be possible in principle, but certainly cannot be considered as practical. Accordingly, we have started an experimental and numerical investigation regarding the feasibility of R2-DQF MAS NMR experiments on dilute spin-1/2 isotopes. An organotin compound, [(chex3Sn)2S], serves as our model sample, containing two crystallographically inequivalent Sn sites per molecule; the isotope 119Sn has a natural abundance of slightly less than 10 %. Preliminary 119Sn R2-DQF MAS NMR experiments on this sample were successful (Fig. 3.7-8) and indicate that in the future similar experiments may also become possible on 29Si in natural abundance.
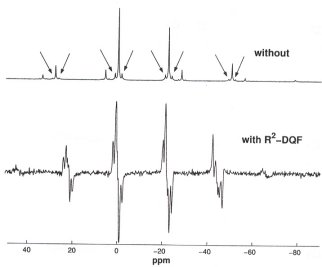 |
Disorder in solid Si(SnMe3)4 and Ge(SnMe3)4 (X. Helluy and A. Sebald; in collaboration with P. Bernatowicz/Warsaw and R. Dinnebier/Bayreuth)
This investigation is a continuation of a long-term project, aiming to characterize the structural and dynamic solid-state properties of crystalline phases of a range of molecular solids, consisting of small, non-polar molecules of the type M'(EMe3)4 with E,M' = C,Si,Sn. For instance, our previous investigations on C(SiMe3)4 and Si(SiMe3)4 have demonstrated that these two compounds exist as orientationally ordered low-temperature phases, both displaying rich and different molecular reorientational dynamics. In contrast, crystalline C(SnMe3)4 was found to represent a dynamically five-fold disordered phase over a wide range of temperatures (see Annual Report 1999). Yet another set of solid-state properties is found for the two (isostructural) compounds Si(SnMe3)4 and Ge(SnMe3)4. Combined constraints from high resolution X-ray powder diffraction measurements and variable-temperature magic-angle-spinning 13C, 29Si, and 119Sn NMR experiments demonstrate low crystallographic and molecular symmetries for the low-temperature phases of Ge(SnMe3)4 and Si(SnMe3)4 at temperatures T < 325 K. The (minor) temperature effects observed in 119Sn and 13C CP MAS NMR spectra of these compounds (Figure 3.7-9) are predominantly due to the intrinsic temperature dependencies of chemical shielding, they are not caused by molecular dynamics or structural phase transitions. These experimental NMR data further illustrate that safe assignment of molecular symmetry from spectrally resolved multiplicities of resonances needs to take the intrinsic temperature dependencies of chemical shielding into account in order to avoid wrong interpretations, in particular overestimation of molecular symmetry.
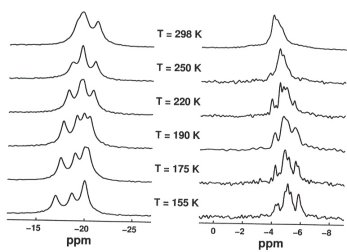 |

Tel: +49-(0) 921 55 3700 / 3766, Fax: +49-(0) 921 55 3769, E-mail: bayerisches.geoinstitut(at)uni-bayreuth.de