

 Previous page
Previous page
Numerically exact lineshape simulations of double-quantum-filtered MAS NMR spectra (S. Dusold and A. Sebald)
We have recently demonstrated for a number of small isolated dipolar coupled spin systems that numerically exact spectral lineshape simulations are feasible for the determination of all parameters describing the spin system, provided that efficient numerical methods are employed. Applying these lineshape-simulation approaches to MAS NMR (magic-angle spinning nuclear magnetic resonance) spectra of spin systems containing more than two coupled spins-1/2 in principle permits the determination of the complete structure of an entire molecule or a molecular fragment from straightforward one-dimensional MAS NMR spectra. However, in many practical applications, the presence of numerous additional resonances in the MAS NMR spectra will severely hamper any lineshape-simulation procedures. For such cases it is necessary to suppress unwanted (background) resonances and to selectively filter out only those resonances of interest. Spectral filtering can be achieved by various so-called double-quantum-filtration (DQF) pulse sequences. DQF is demonstrated in Figure 3.7-10a for 13C MAS NMR spectra of a mixture of Na-pyruvate with 13C in natural abundance (90%) and 2,3-13C2-labelled Na-pyruvate (10%): the conventional 13C MAS NMR spectrum (top) displays all 13C resonances, after DQF the 13C MAS NMR spectrum (bottom) only shows the resonances of the 2,3-13C2-labelled spin pair. In order to extract unknown parameters from the lineshapes in these DQF MAS NMR spectra, it is necessary to include the effects of the entire pulse sequence in the simulations. That it is possible to simulate DQF MAS NMR spectra very accurately is shown in Figure 3.7-10b, where an experimental (top) DQF MAS NMR spectrum is compared to the corresponding simulated spectrum (bottom). We expect these and similar methods to become useful MAS NMR approaches for the accurate determination of internuclear distances in a wide variety of spin systems, including 29Si and 31P in inorganic silicates and phosphates.
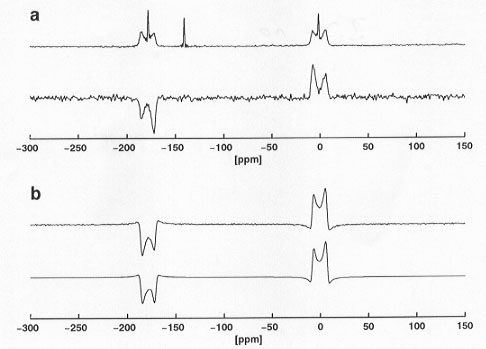 |
Fig. 3.7-10: a) conventional (top) and double-quantum-filtered (bottom) 13C MAS NMR spectra of a mixture of Na-pyruvate with 13C in natural abundance (90%) and 2,3-13C2-labelled Na-pyruvate (10%); b) comparison of an experimental (top) and a simulated (bottom) DQF 13C MAS NMR spectrum of 2,3-13C2-labelled Na-pyruvate. |
Disorder in solid C(SnMe3)4 (X. Helluy, J. Kümmerlen and A. Sebald, in collaboration with P. Bernatowicz and R. Dinnebier/Bayreuth)
Previous single-crystal X-ray diffraction experiments on C(SnMe3)4 have indicated the presence of a pseudo-fivefold disorder in this material. Diffraction techniques alone can not distinguish between static and dynamic orientational disorder. We have employed one- and two-dimensional 119Sn NMR techniques to investigate further the disorder in solid C(SnMe3)4. Variable-temperature 119Sn NMR experiments in the temperature range T = 150-290 K positively identify the dynamic nature of the pseudo-fivefold disorder by exploiting information encoded in the 119Sn chemical shielding tensor. Amongst numerous different possible dynamic-disorder models, a model where each tin atom in the C(SnMe3)4 molecule occupies the twenty corners of a dodecahedron with equal probability agrees best with the combined experimental one- and two-dimensional 119Sn NMR evidence. This is illustrated in Figure 3.7-11, where the twenty-fold exchange over the corners of the dodecahedron is illustrated schematically (Fig. 3.7-11a). Note that this mode of molecular reorientation requires the simultaneous occurrence of small- and large-angle reorientational jumps of the C(SnMe3)4 molecule. In Figure 3.7-11b the contour plot of a low-temperature two-dimensional 119Sn exchange experiment is shown in comparison with the best-fit calculated spectrum according to the twenty-fold exchange model. The combined exchange-rate constants from all variable-temperature one- and two-dimensional 119Sn NMR experiments yield an activation energy Ea 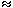 32 kJmol-1 for the twenty-fold exchange process in solid C(SnMe3)4.
32 kJmol-1 for the twenty-fold exchange process in solid C(SnMe3)4.
 |
Fig. 3.7-11: a) pictorial representation of the dynamic disorder model in solid C(SnMe3)4; b) contour plots of 119Sn two-dimensional exchange NMR spectra under non-spinning conditions at T = 150 K with short mixing time (  m = 5 ms); left: experimental spectrum, right: best-fit simulated spectrum.
m = 5 ms); left: experimental spectrum, right: best-fit simulated spectrum. |
The pseudo-cubic phases of SiP2O7 and TiP2O7 as seen by various 31P MAS NMR experiments (X. Helluy and A. Sebald, in collaboration with C. Marichal/Mulhouse)
Spin-diffusion based 31P MAS NMR investigations (see Annual Report 1997) on the pseudo-cubic phase of SiP2O7 have now been completed and include full characterization of the isostructural phase TiP2O7 as well. The combined results may be summarized as follows:
- various 31P MAS NMR techniques fully and independently confirm for both phases the cubic space group
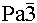 with a 3 x 3 x 3 superstructure as the correct choice, as had been previously proposed from refinement of X-ray diffraction data of both phases;
with a 3 x 3 x 3 superstructure as the correct choice, as had been previously proposed from refinement of X-ray diffraction data of both phases; - a simple comparison of one-dimensional 31P MAS NMR spectra of the two isostructural phases (see Fig. 3.7-12) demonstrates that common qualitative, isotropic 31P chemical-shielding-based assignment schemes to identify 31P resonances with individual P sites in the structure are not generally applicable to condensed phosphate systems;
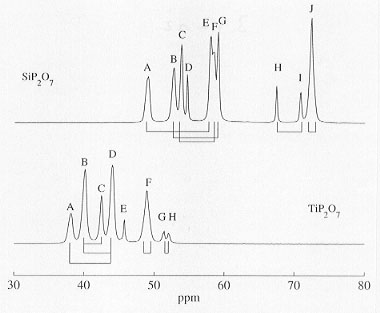 |
Fig. 3.7-12: Comparison of 31P MAS NMR spectra of SiP2O7 (top) and TiP2O7 (bottom). Also indicated are the pairwise P-O-P connectivities as determined by J-coupling connectivities; resonances D (SiP2O7) and E (TiP2O7) correspond to special P-O-P sites with magnetically equivalent intra-P2O7 31P spin pairs and hence do not display the corresponding intra-P2O7 31P J-coupling connectivity. |
- constraints from J-coupling-based ('through-bond') 31P NMR experiments successfully identify P-O-P subunits in the structures and thus are extremely useful for the reduction of the originally very large number of possible assignment permutations;
- full and unambiguous assignment requires additional dipolar-coupling-based ('through-space') 31P NMR experiments. So-called single-quantum - double-quantum 31P correlation MAS NMR experiments yield unique assignment for both phases. So-called zero-quantum ('spin diffusion') 31P MAS NMR experiments, however, are successful for spectral assignment only for SiP2O7 but fail for TiP2O7, owing to the lesser 31P NMR spectral resolution as compared to SiP2O7.
The final complete assignment of the 31P resonances to the crystallographic P sites P1 to P11 in the structures of SiP2O7 and TiP2O7 is given below (compare Fig. 3.7-12):
| P site: | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 |
| 31P resonances: TiP2O7 SiP2O7 |
A |
B |
F |
F |
B |
D |
C |
D |
H |
E |
G |
| A | B | J | J | C | F | G | E | I | D | H |
MAS NMR techniques for the determination of molecular conformations (M. Bechmann, S. Dusold and A. Sebald, in collaboration with M. Stumber, U. Haeberlen/Heidelberg, H. Förster/Karlsruhe, T. Lis/Wroclaw and J.N.S. Evans/Washington)
The recently started, extended joint project (see Annual Report 1998), aiming to develop and apply MAS NMR techniques for the direct determination of molecular conformations and focussing on the biochemically important phosphoenol-pyruvate (PEP) moiety has accomplished some of its initial tasks. First, the orientations of all 31P and 13C chemical shielding tensors in a suitable PEP-model compound of known structure had to be determined. (NH4)3(PEP) was chosen for this purpose. From 31P single-crystal NMR studies and 13C-31P heteronuclear dipolar recoupling MAS NMR experiments on (NH4)3(PEP) with 13C in natural abundance, the absolute orientation of the 31P and the 13C2 chemical shielding tensors in the molecular frame of the PEP moiety are now known (see Fig. 3.7-13). In order to determine the 13C1 and 13C3 chemical shielding tensor orientations, it is necessary to carry out 13C MAS NMR experiments on fully 13C-enriched U-13C-(NH4)3(PEP): the remaining unknown orientational parameters of the 13C spin systems are encoded in the lineshapes of various different rotational-resonance 13C MAS NMR spectra (see Fig. 3.7-14). Once this data analysis is complete, the first stage of the project is complete. The following phase will be concerned with the selection and testing of suitable MAS NMR experiments to correlate the 13C1-13C2 chemical shielding tensors.
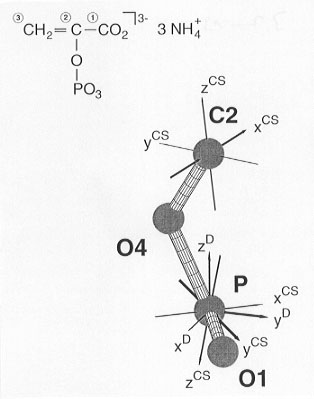 |
Fig. 3.7-13: The orientation of the 31P and 13C2 chemical shielding tensors in the molecular frame of (NH4)3(PEP). |
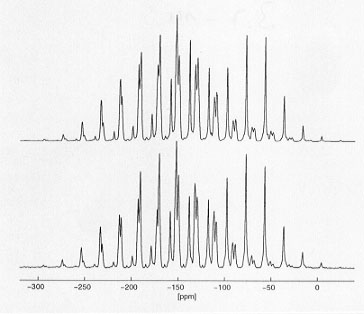 |
Fig. 3.7-14: Comparison of an experimental (bottom) and a best-fit calculated (top) 13C MAS NMR spectrum of U-13C-(NH4)3(PEP). |

Tel: +49-(0) 921 55 3700 / 3766, Fax: +49-(0) 921 55 3769, E-mail: bayerisches.geoinstitut(at)uni-bayreuth.de