

 Previous page
Previous page
The presence of dissolved water in many minerals is known to have a significant effect on their physical, mechanical and chemical behavior. Thus, it is important to understand the mechanisms of incorporation or loss of water-derived species in the major minerals of the Earth's interior. Experimental studies have demonstrated that water-derived species diffuse very rapidly in a range of nominally anhydrous silicate minerals, including olivine, clinopyroxene, orthopyroxene and garnet. However, under the conditions of the experiments, the mechanism of diffusion in these iron-bearing minerals generally involves a cooperative exchange process of protons with the excess charge on iron ions in octahedral cation sites (polarons). Essentially, protons move into (or out of) the mineral by decreasing (or increasing) the polaron concentration. This process may drive the hydrogen content of the mineral towards equilibrium but the polaron concentration (essentially the ferric/ferrous ratio of the mineral) away from equilibrium. At higher temperatures and/or longer times, hydrogen will be incorporated in these minerals as defect associates, such as protonated magnesium or silicon vacancies, allowing equilibrium of all defects with the imposed thermochemical environment. Such incorporation will occur for iron-free as well as iron-bearing minerals.
The present study, which is based on partial hydration or dehydration experiments and FTIR analysis, aims to constrain more fully the speciation of the mobile OH defects in olivine and rates of diffusion at a variety of pressure and temperature conditions. Diffusion of OH defects in single crystals of olivine is being investigated for diffusion parallel to the [100], [010] and [001] crystallographic axes during both dehydration and hydration. We use xenolithic crystals from San Carlos, Arizona, that have no cracks or inclusions. The dehydration experiments are performed in a room-pressure furnace at temperatures between 800º and 1200ºC, with the oxygen fugacity controlled using mixed CO and CO2 gases. The hydration experiments are performed at temperatures between 800º and 1200ºC and a fluid (H2O) pressure of 1 GPa in a piston-cylinder apparatus or 300 MPa in an internally heated, gas-medium pressure vessel. For both apparatus, we place the samples inside platinum-rhodium capsules along with 10 mg of water and a mixture of olivine (90%) and orthopyroxene (10%) powder. In order to control the oxygen fugacity at Ni/NiO (or Fe/FeO) during the experiments, NiO (or FeO) powder and Ni (or Fe) foil are also included in the capsule prior to welding.
The hydroxyl distributions within our samples are analyzed using polarized FTIR spectroscopy. Diffusion profiles for the OH defect species are measured parallel to [100], [010] and [001] for each sample. An example of the distribution of OH in a sample treated hydrothermally at a fluid pressure of 300 MPa for 1 h at 850ºC, with the oxygen fugacity buffered at Fe/FeO is given in Figure 3.4-4. Under these conditions, diffusion occurs predominantly through redox exchange between protons and polarons. The defect diffusivity
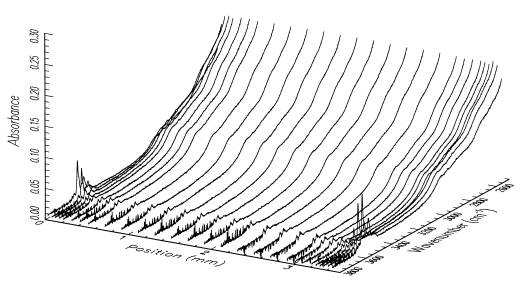 |
is obtained by fitting a solution of a theoretical diffusion law to the OH concentrations, which are determined by integration of the infrared bands between 3650 and 3400 cm-1. Our current results indicate that incorporation of OH species into olivine is a 2-step process with an initial stage of proton-polaron exchange, followed by a slower diffusion of OH species, probably as defect associates of protons and octahedral cation vacancies. Diffusivities measured for the initial exchange are anisotropic, with diffusion being fastest parallel to [100]. Diffusivities measured during the slower incorporation process are also anisotropic, but with diffusion fastest parallel to [001]. The absolute diffusivities for this second stage are similar to those reported for the diffusion of octahedral cation vacancies. It is also interesting to note that only after this latter stage is complete can the samples attain equilibrium with the imposed hydrothermal environment.

Tel: +49-(0) 921 55 3700 / 3766, Fax: +49-(0) 921 55 3769, E-mail: bayerisches.geoinstitut(at)uni-bayreuth.de